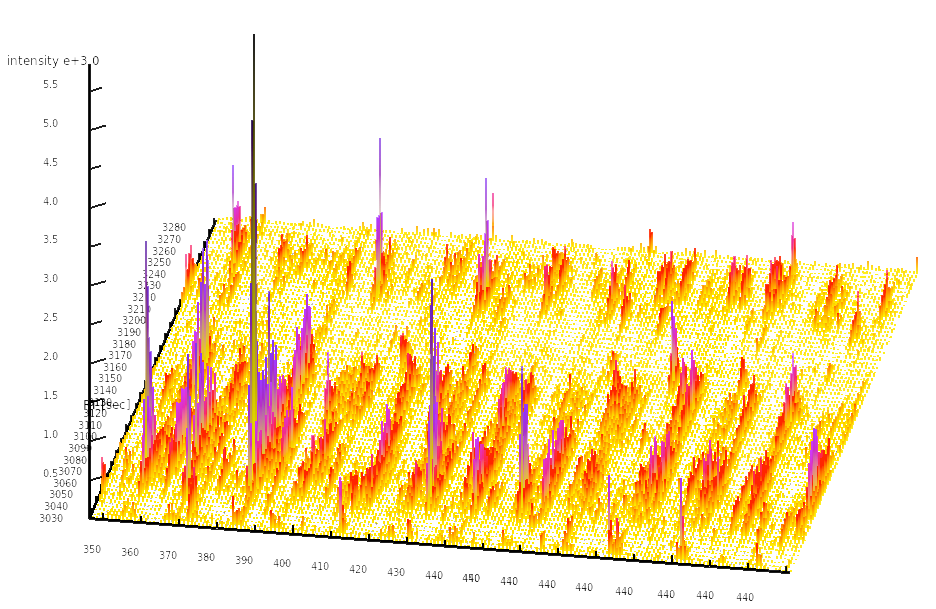
Mass-spectrometry based quantitative proteomics allows researchers to
accurately quantify the dynamics of protein abundance and protein activity in
biological systems. In order to increase the quantitative accuracy and the
throughput of proteomics methods, we have developed a novel targeted proteomics
method called SWATH-MS that is based on data-independent acquisition (DIA)
which aims to complement traditional mass spectrometry-based proteomics
techniques such as shotgun and SRM methods. In principal, it allows a complete
and permanent recording of all fragment ions of all peptide precursors in a
biological sample and can thus potentially combine the advantages of shotgun
(high throughput) with those of SRM (high reproducibility and sensitivity).
To analyze the SWATH-MS data, we developed OpenSWATH,
an automated software to perform targeted data extraction from the SWATH-MS
maps. Our software allows to perform automated data extraction, peak-picking
and feature-detection in chromatographic traces, thus performing a complete
SWATH-MS data analysis completely automatically; the only input are the raw
MS/MS files as well as a transition library to perform the targeted data
extraction. After feature detection, we use the mProphet
algorithm for error rate estimation.
Using SWATH-MS in conjunction with OpenSWATH, we have successfully quantified
over 900 proteins in the pathogen Streptococcus pyogenes in a single
LC-MS/MS injection (more than any previous study), allowing us to study the
response of the pathogen to human blood plasma in unprecedented detail. We also
could quantify over 1900 human proteins in an AP-MS pulldown experiment and
identify over 500 high-confidence physical protein-protein interactions of the
14-3-3β scaffold protein, giving us direct insight into the dynamics of a large
protein interaction network.
Relevant publications:
Röst HL, Liu Y, D'Agostino G, Zanella M, Navarro P, Rosenberger
G, Collins BC, Gillet L, Testa G, Malmström L, Aebersold R.
TRIC: an automated alignment strategy for reproducible protein
quantification in targeted proteomics.
Nat Methods. 2016 Sep;13(9):777-83.
Guo T, Kouvonen T, Koh CC, Gillet LC, Wolski WE, Röst HL,
Rosenberger G, Collins BC, Blum LC, Gillessen S, Joerger M, Jochum W,
Aebersold R.
Rapid mass spectrometric conversion of tissue biopsy samples into permanent quantitative digital proteome maps. Nature Medicine.
2015 Apr;21(4):407-13.
Rosenberger G, Koh CC, Guo T, Röst HL, Kouvonen P, Collins BC,
Heusel M, Liu Y, Caron E, Vichalkovski A, Faini M, Schubert OT, Faridi P,
Ebhardt HA, Matondo M, Lam H, Bader SL, Campbell DS, Deutsch EW, Moritz RL,
Tate S, Aebersold R.
A repository of assays to quantify 10,000 human proteins by SWATH-MS. Scientific Data. 2014. Sept 16.
Röst HL, Rosenberger G, Navarro P, Gillet L, Miladinović SM, Schubert OT, Wolski W, Collins BC, Malmström J, Malmström L, Aebersold R. OpenSWATH enables automated, targeted analysis of data-independent acquisition MS data. Nat Biotechnol. 2014 Mar 10;32(3):219-23. doi: 10.1038/nbt.2841.
Collins BC, Gillet LC, Rosenberger G, Röst HL, Vichalkovski A, Gstaiger M, Aebersold R. Quantifying protein interaction dynamics by SWATH mass spectrometry: application to the 14-3-3 system. Nat Methods. 2013 Dec;10(12):1246-53. doi: 10.1038/nmeth.2703.
Gillet LC, Navarro P, Tate S, Röst H, Selevsek N, Reiter L, Bonner R, Aebersold R. Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis. Mol Cell Proteomics. 2012 Jun;11(6):O111.016717. doi: 10.1074/mcp.O111.016717.
A first step towards a better understanding of molecular systems is to study
the reaction of a system to perturbations and infer basic internal causal
processes from these studies. Possible perturbations can be environmental (by
subjecting an organism to different stress conditions) or they can be genetic
(e.g. mutations) that may be induced or natural. The latter approach can lead to a
direct, causal understanding of how certain genetic features influence the
molecular phenotype of a cell and then determine the phenotype of an organism
on a macroscopic level. Using such perturbation data, researchers have
successfully uncovered direct relationships between genetic features and
transcript (eQTL) or protein (pQTL) abundance. In addition, multiple genetic
regions have been linked to certain macroscopic phenotypes (such as disease
phenotypes) using genome-wide association studies (GWAS) in humans. Finally,
for medical applications and diagnostic purposes it is interesting to find
so-called protein "biomarkers" that directly relate the abundance of a protein
to a (disease) phenotype.
Rapid advances in pQTL, GWAS and protein biomarker studies have been reported
in recent years which rely on technological breakthroughs in the fields of
genetic sequencing and protein quantification. Currently (computational)
proteomics directly improves accuracy and reliability of biomarker and pQTL
studies by improved identification and quantification results (see section
above). However, it is still an open question how to combine these individual
glimpses of a biological system into a consistent and functional understanding
of the system.
We plan to study genotype to phenotype relations using clinical isolates of a
model pathogen, Streptococcus pyogenes. We plan to investigate the
relationship between genetic point mutations and observed protein quantities in
each strain. We plan to use SWATH-MS to obtain high coverage and consistent
quantification over multiple samples in a targeted proteomics fashion with high
throughput. From this we hope to gain novel insights into the interplay of
genetic adaptation and transcriptional and translational regulation of S.
pyogenes and, finally, how this affects the virulence phenotype of
individual strains.
Relevant publications:
Röst HL, Malmström L, Aebersold R.
Reproducible quantitative proteotype data matrices for systems biology.Mol Biol Cell. 2015 Nov
Guo T, Kouvonen T, Koh CC, Gillet LC, Wolski WE, Röst HL,
Rosenberger G, Collins BC, Blum LC, Gillessen S, Joerger M, Jochum W,
Aebersold R.
Rapid mass spectrometric conversion of tissue biopsy samples into permanent quantitative digital proteome maps. Nature Medicine.
2015 Apr;21(4):407-13.
Röst HL, Rosenberger G, Navarro P, Gillet L, Miladinović SM, Schubert OT, Wolski W, Collins BC, Malmström J, Malmström L, Aebersold R. OpenSWATH enables automated, targeted analysis of data-independent acquisition MS data. Nat Biotechnol. 2014 Mar 10;32(3):219-23. doi: 10.1038/nbt.2841.
Simulation studies
Often, biological phenomena cannot be studied or measured directly (or doing so
would be too resource-intensive) and researchers need to use in silico
simulations to analyze complex phenomena. Some well-known examples in
computational biology include protein folding or kinetic modelling where
computer simulation-based approaches are used heavily. In mass-spectrometry
based proteomics, peptide digestion, chromatographic separation and
collision-induced dissociation to fragment charged peptide precursor ions are
complex phenomena which can be approached using simulation to provide
predictions and insights into error rates during peptide identification and
peptide quantification.
Our software, the SRMCollider, allows to model all individual steps in a
LC-MS/MS experiment (digestions, chromatographic separation, fragmentation),
specifically taking into account the challenges of targeted proteomic where
only a few fragment ions are monitored for each peptide. This allowed us to
investigate the question of assay redundancy in SRM and SWATH-MS experiments
and make concrete predictions about assay specificity in a targeted proteomics
setting. We have successfully applied these simulations for multiple studies in
the Aebersold lab, including proteomes as diverse as Mycobacterium
tuberculosis, Saccharomyces cerevisiae and Homo sapiens.
Relevant publications:
Schubert OT, Mouritsen J, Ludwig C, Röst HL, Rosenberger G, Arthur PK, Claassen M, Campbell DS, Sun Z, Farrah T, Gengenbacher M, Maiolica A, Kaufmann SH, Moritz RL, Aebersold R. The Mtb proteome library: a resource of assays to quantify the complete proteome of Mycobacterium tuberculosis. Cell Host Microbe. 2013 May 15;13(5):602-12. doi: 10.1016/j.chom.2013.04.008.
Picotti P*, Clément-Ziza M*, Lam H*, Campbell DS, Schmidt A, Deutsch EW, Röst H, Sun Z, Rinner O, Reiter L, Shen Q, Michaelson JJ, Frei A, Alberti S, Kusebauch U, Wollscheid B, Moritz RL, Beyer A, Aebersold R. A complete mass-spectrometric map of the yeast proteome applied to quantitative trait analysis. Nature. 2013 Feb 14;494(7436):266-70. doi: 10.1038/nature11835.
Hüttenhain R, Soste M, Selevsek N, Röst H, Sethi A, Carapito C, Farrah T, Deutsch EW, Kusebauch U, Moritz RL, Niméus-Malmström E, Rinner O, Aebersold R. Reproducible quantification of cancer-associated proteins in body fluids using targeted proteomics. Sci Transl Med. 2012 Jul 11;4(142):142ra94. doi: 10.1126/scitranslmed.3003989.
Röst H, Malmström L, Aebersold R. A computational tool to detect and avoid redundancy in selected reaction monitoring. Mol Cell Proteomics. 2012 Aug;11(8):540-9. doi: 10.1074/mcp.M111.013045.
Gillet LC, Navarro P, Tate S, Röst H, Selevsek N, Reiter L, Bonner R, Aebersold R. Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis. Mol Cell Proteomics. 2012 Jun;11(6):O111.016717. doi: 10.1074/mcp.O111.016717.